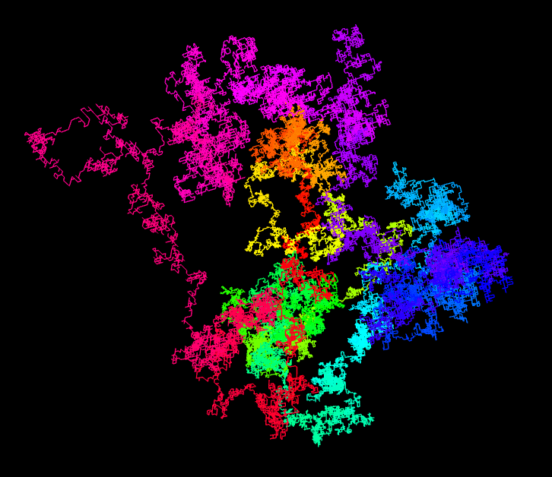
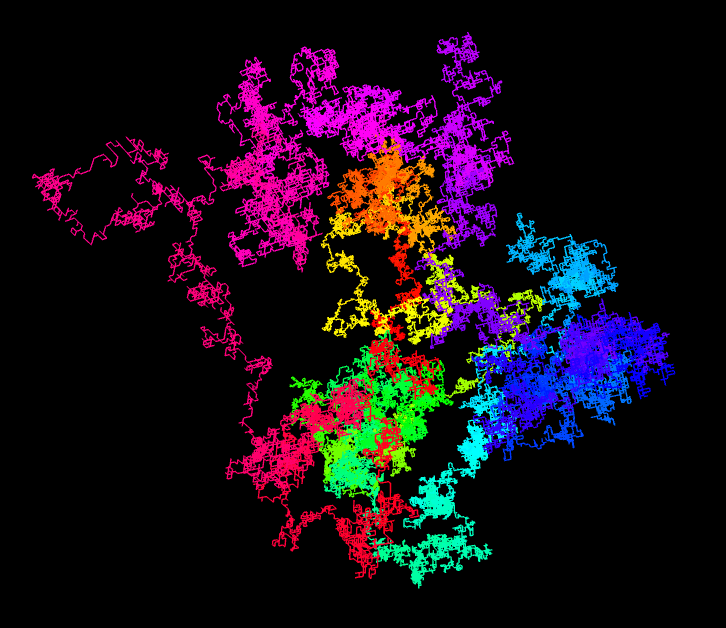
Inspired by a web page with random walks based on π, I implemented this idea with the 3D engine Babylon.js. You can look at the result on this page. When opening the link, you see the script on the left, on the right you can zoom and rotate the resulting 3D structure. The first 100’000 decimal digits of π are used to generate the 3D random walk. A 0 encodes for a step in the +x direction, a 1 for a step in the -x direction. The digits 2 and 3 are used to move along the y-axis and the digits 4 and 5 for the z-axis. The remaining digits 6 to 9 are simply ignored.
The resulting 3D model resembles the protein structures as reconstructed from measurements performed with the X-ray detectors I help to develop. The corresponding discipline, called macromolecular crystallography, is the main application of our large area detectors. An example of a protein structure is shown in the image below.
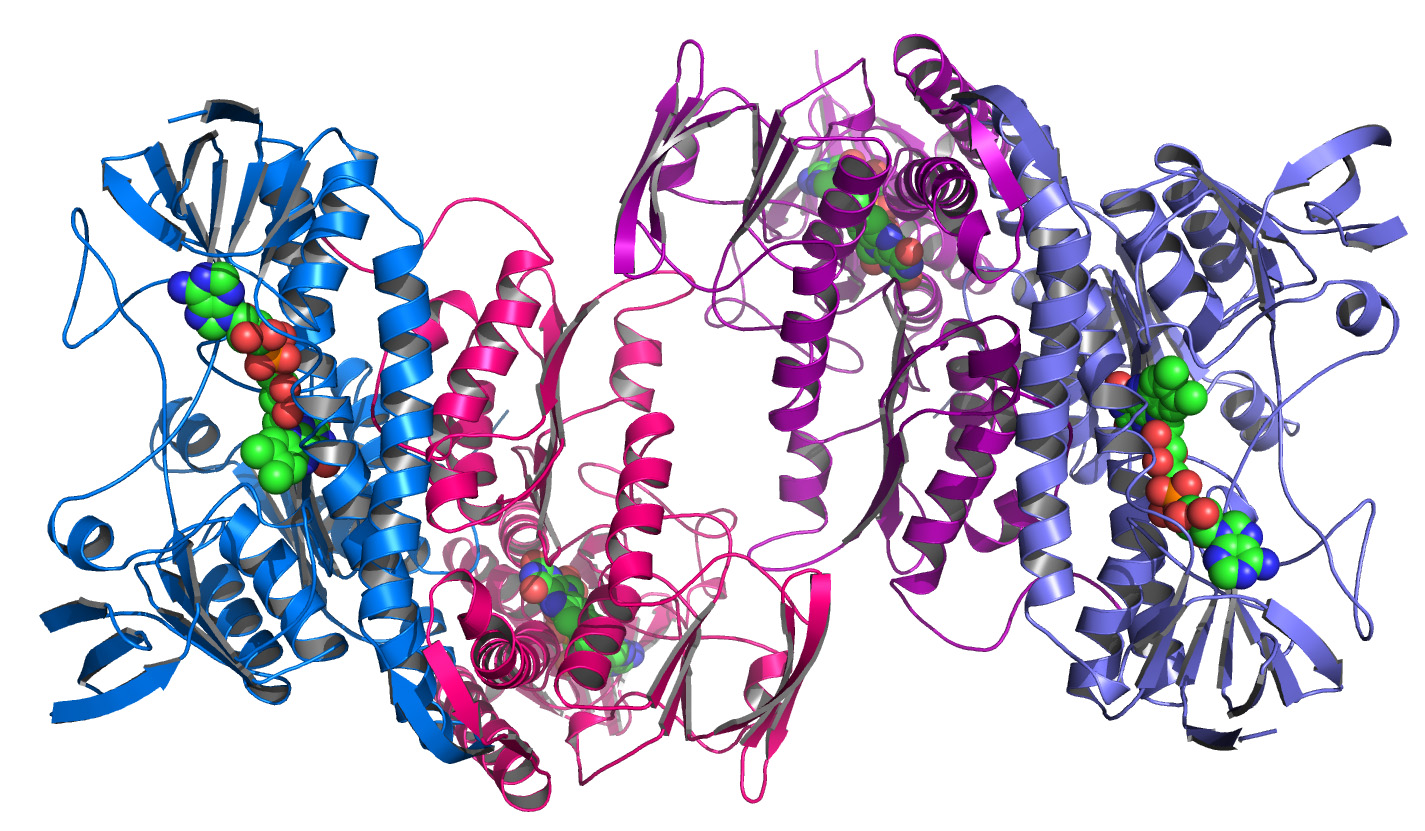

To determine the three dimensional structure of a protein, first a crystal of the corresponding substance has to be grown. In a second step this crystal is exposed to brilliant X-ray radiation, for example at a Synchrotron beam line. An X-ray detector is used to record the resulting diffraction pattern, which is a highly symmetric pattern of Bragg peaks. Finally, after a Fourier transform and after solving the phase problem, an electron density map of the protein is obtained.
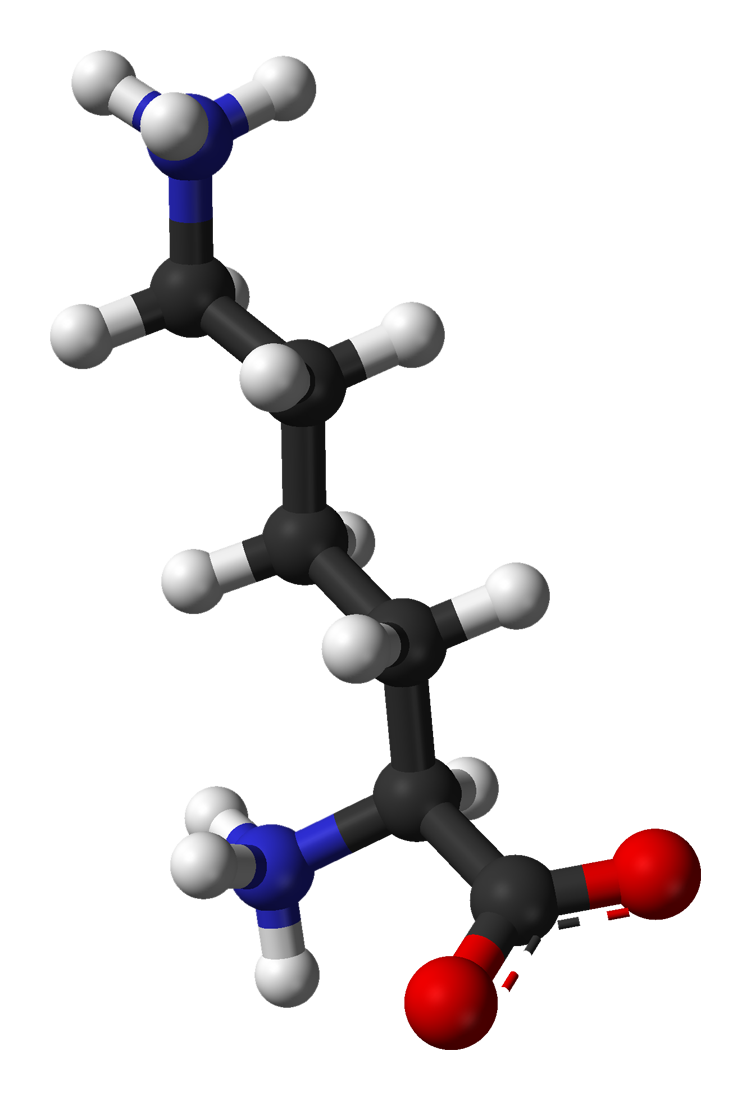
Proteins are fascinating structures assembled from hundreds of left-handed amino acids molecules. The amino acid alphabet as encoded in the DNA encompasses 20 different basic building blocks. For a protein consisting of 100 amino acids, there are 10130 different ways to arrange them into a sequence. Out of this huge number of possible proteins, only a tiny fraction has a biological activity (Taylor et al. estimate e.g. a fraction of 10-23). After 50 years of research, there is still no evidence how these proteins could have evolved de novo from a racemic mixture of amino acids.




Comments by Pipi